
一切來自於某人聽說發生在某間醫院的某位科主任研究室內的小故事:
某醫生:「我手上有某個疾病的 30 個病人樣本要做全基因體分析,找出導致這個疾病的致病基因和相關的變異點。你們收多少錢?」
A 廠商:「某醫師,這是我們的報價單,列出 30 個 WGS 定序的價錢,這是目前最具市場競爭力的價錢,要買要快,產能最近都滿載,要排隊喔。另外,我們還免費贈送“標準的分析”喔!」
半年後 …
B 廠商:「我們是 XX 基因科技公司,提供各種 NGS 的服務,包含專業的生物資訊服務,不知道您有沒有這方向的需求?」
某醫生:「你們這些 NGS 廠商都在亂搞,半年前我花了幾百萬做出來的資料根本沒用!」
B 廠商:「怎麼會呢?請問您是跑了什麼 Pipeline 呢?」
某醫生:「『你們』廠商提供的標準分析呀!Standard Analysis!」
B 廠商:「那『他們』有說是使用那些軟體嗎?」
某醫生:「就說是標準分析了,『你們』應該要知道才是!怎麼還問我呢!我就說『你們』這些廠商都在亂搞嘛!NGS 根本沒用嘛!」
本文的宗旨就是希望能讓讀者對「標準分析流程」有基本認識,進而了解標準分析流程的限制與不足,最後再給一些實用的建議,讓大家都能變成「巷子內」的專家。
因為要看變異點資訊,所以「標準分析」就是指 Resequencing,步驟如下:

大部分廠商所謂的「標準分析」主要包含 Read Mapping 和 Variant Calling 這 2 個步驟(即所謂的二級分析),有些廠商會再多提供一些常用基因資料庫的 Annotation(即三級分析),也看過有廠商的「標準分析」是指 FASTQ 的 QC Report 而己(即一級分析)。所以這邊想要分享的心得是:
所謂「標準分析」的定義並沒有一定的標準
大家心中可能在問到底有多少級分析呀?目前比較有共識的定義可以參考下圖:

簡單的分辨方法是,若最後廠商交付的只有 FASTQ 檔案,就是只做到一級分析(Primary Analysis);若是交付 VCF 檔案,則一定可以確定有做到二級分析 (Secondary Analysis)或再加部分的三級分析。
若只有提供二級分析,雖然離大家期待的最終結果或報告還有一段距離,但若在二級分析沒做好的情況下,要不就少了關鍵的變異點資訊,要不就多了很多假的變異點資料,這些都會導致最後分析不出結果或做出錯的結論。所以,我們事先應該要釐清的問題是:
這個 VCF 是怎麼做出來的?
通常一級分析主要是確認 Read 數量是否足夠以及 Read Quality 來判斷這次上機所產生資料的品質,經常使用的工具是FastQC。而二級分析時,會在 Read Mapping 完後看看 unmapped reads 的比例和 mapping coverage 來確認這些出來的資料是否完整以及是否有被污染; 另外 Variant Calling 後也會看產生 VCF 的變異點個數來大概確認資料是否有出問題,例如 ATCG 比例、Homo和 Hetro的比例等。
而 Variant Calling 這邊會因為使用不同的 caller 而有不同的結果,詳細的介紹請參考:
光 SNP和 Indel 的 variant callers 就這麼精彩了,其實還有更精彩且更複雜的戰場是在 Structural Variant 的偵測。為什麼 Structural Variant Detection 會有這麼複雜呢?接下來就進入本文的內容重點。
Resequencing 這種 Mapping-based Pipeline 主要適用於這個物種基因體單純,個體變異差異較小的情況,可以利用 Reference Genome 當參考依據,以 Read Mapping 的方式快速找到最合適的位置,大大減少運算量(相對於 Assembly-based 方式而言)。Reference 不完整或個體變異性大都會影響 Resequencing 的效果,所以人類到底合不合適 Resequencing 的分析流程呢?這個問題最早在 2012 年 Heng Li 大大的 Fermi 論文中就提到這種 Mapping-based approach 有先天上的缺陷,他在文中是用「 Fundamental Flaw」來描述。

為什麼 Heng Li 有資格對 Resequencing 給出這麼嚴厲的批評呢?因為 Read Mapping 中最著名的工具 BWA 是他寫的,也因此他最清楚 NGS 的短序列在做 Read Mapping 時會遭遇到那些問題與挑戰。所以,本文統整出四件事大家必須要先了解:
第一、Human Reference Genome 目前仍不完整
Human Reference Genome 從西元 2000 年 5 月 Draft 公佈 HG1 開始進版,到 2009 年的 HG19,之後開始以上 patch 的方式來做修正,到 2013 年 1 月的 p13 後,當年 12 月直接改版到 HG38,之後一樣以上 patch 的方式來修正,目前最新版本為 p13 。 為什麼要像軟體一樣一直更新呢?說穿了,跟軟體更新一樣:「修bug!」但為什麼會有 bug 呢?以下圖為例,目前 Human Reference Genome 中約有 5.3% 是以「 N 」來表示,奇怪了?DNA 不是以 ATCG 等四種核甘酸所組成,什麼時候多出 N 來了呢?N 就是不確定它到底是 ATCG 那一個,只確定那邊有 DNA 存在,這些 N 大部分發生在 Centromere 和 Telomere 上(如圖上所標示的藍色區塊),但也有一堆小片段的 N 散落在基因體的很多地方(下圖並未表示),也就是目前的 Reference Genome 遍佈著 N,導致在做 Read Mapping 會遇到不少挑戰。
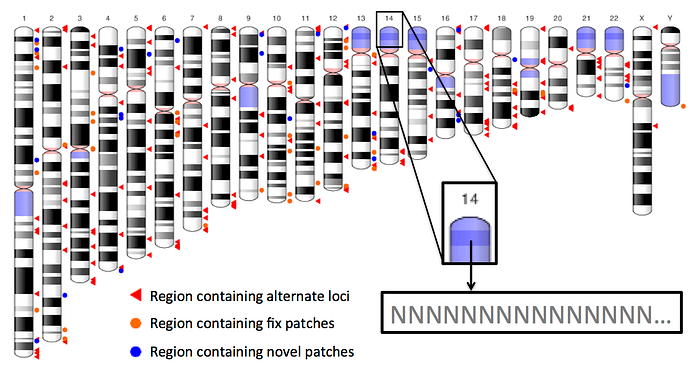
第二、尚有很多的 DNA 序列不在 Reference Genome 中
中研院生醫所郭沛恩所長今年發表在 Nature Communications 的文章利用 The 1000 Genomes Project 的 26 個人種中發現約有 60Mbps 不在目前的 Reference Genome 中,這些有可能是出現在 N-Gap 中,也有可能不是。但不管是或不是,唯一確定的是,這些都會影響到 Read Mapping,因為這些 DNA 片段並不在 Reference 中,又因為 NGS 短序列的特性,很容易就誤貼到不屬於它原本位置的地方,也因為導致接下來的 Variant Calling 變得這麼難判斷。
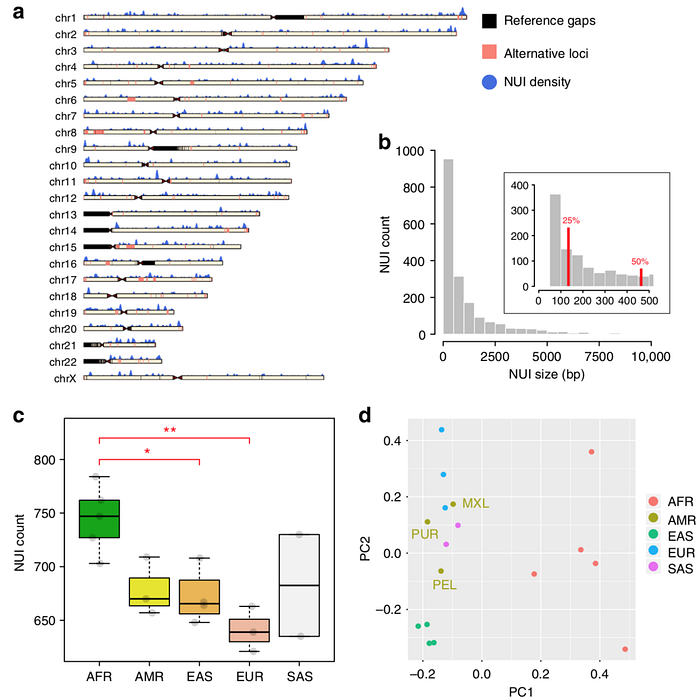
第三、用單一的 Reference 來描述世界上的每一個人並不合適
目前 Resequencing 的作法是假設人類這個物種的基因體應該是大同小異,所以先建立出可以描述人類基因體的 Reference Genome 樣版後,每個人就可以跟 Reference 來比較差異,找出每個變異點 (Variant)後即可研究基因體與疾病的關係。但問題是:
用同一個 Reference Genome 來描述全部人類是否合適呢?
一樣以 The 1000 Genomes Project 為例,分析全球26個人種在 SNP 獨特性的統計圖,其中每個圓餅圖代表一個人種,圓餅圖的大小代表找到 SNP 的數量。而 SNP 就是跟 Reference Genome 不一樣的地方,很明顯看出目前 Reference 跟非洲人種比較不像,所以非洲人的圓餅圖都偏大。
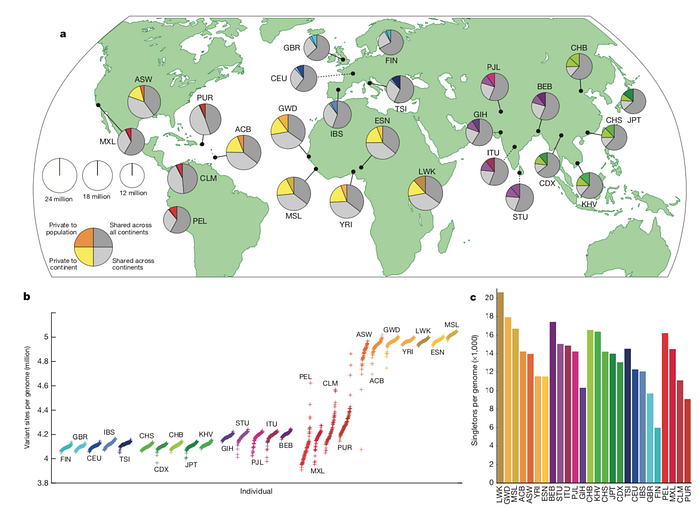
當然,這些數據是建構在不完整的 Reference 下所產生的資料,變異點的正確性還值得進一步探討,但若是用來統計人種間的差異性是沒問題的。
另外,因為目前 Reference 只用一維的方式來表示人類基因體的架構,會造成與實際的狀況差距太大,更何況每個人其實都有兩套染色體,一套來自爸爸,一套來自媽媽,所以 Seven Bridges Genomics 在 2015 年就曾提出 Graph Genome (如下圖),用二維的 Graph 來描述 Population 的分佈,試圖提供更多樣的資訊來表示 Reference Genome。
第四、結構型變異與密集型變異群集
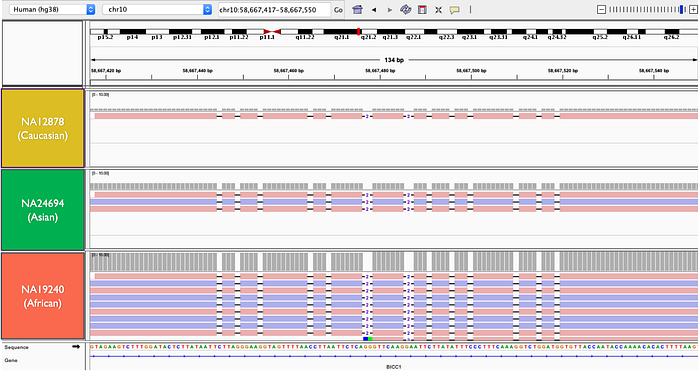
短序列在做 Read Mapping 時,當只有一個單點變異發生時,並不會有什麼技術上的困難。因若有多個單點變異群集或結構型變異時,短序列常常會得到 Soft / Hard-Clipping 的 Alignment 結果,導致後會在做 Variant Calling 時容易出錯,也因此很多 Variant Calling Tools 都會自己再做一下 Local Assembly 來獲得更完整的 Alignment,但遇到 Structural Variant 或密集型變異群集,若導致沒有任何 Read 貼上去,那肯定沒輒了。另外,下圖就是有 13 個 1-bp or 2-bp 的 deletion 所組成的區域,若用短序列,只會看到這邊可能有一個大的 Structural Variant 而己,如同這篇在分析 15,219 個冰島人共同的結構型變異一樣,誤以為這一個區域有結構型變異出現。利用我們新開發出來的 ConnectedReads 分析流程可以完整的呈現出這個區域的真實狀況,如下圖所示,三個不同人種在這個區域有一模一樣的變異群集,這也反應出 Reference Genome 應該要修正才是。
NGS 的確是個好工具,前提是好好地被使用;要如何好好地使用它,首先必須要先多了解它,了解它的能力、了解它的不足、了解它的性情、了解它的美,就能駕馭它、依賴它,進而喜歡上它。
最後,文章提到的論點都是理想面,最後還是得回到現實面,各廠商對品質的要求參差不齊,如何讓大家能得到預期的結果,建議多了解一下廠商所謂的「標準分析」是什麼,例如詢問廠商是跑什麼分析軟體?那個版本?
當然,有些廠商會說:「這些分析軟體都大同小異,所以我們提供免費的標準分析就夠了!」
若真的能得到您想要的結果,那真的恭喜您了!但若沒有得到預期的結果時,建議再回來看看本文,檢視一下分析流程是否合適。
看到好多有趣的基因體資料躺在醫生的抽屜裡好幾年不見天日,因為得不到結果,只因為 …
「免費」的最貴!
